

Copyrights: Vahid Amiri, Mohamadhossein Mohammadi, Mohammad Reza Khosravi Farsani, Arshia Gharehbaghian, Abbas Hajifathali, Zaher Khazaei, Mehdi Allahbakhshian Farsani, 2018. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Introduction: Gene mutation is an infrequent cause of tumor suppressor gene (TSG) defect in de novo AML patients. Instead, it seems that leukemic cells employ epigenetic tricks to attenuate the negative impacts of intact TSGs. Ordinarily, critical TSGs, such as p16INK4A, is hyper-methylated in AML blasts under the impact of master epigenetic regulators, such as UHRF1. In this study, we investigated the correlation between UHRF1 and p16INK4A gene expression levels in newly diagnosed AML patients.
Methods: Bone marrow and peripheral blood samples were obtained from 50 newly diagnosed AML patients and 18 healthy normal control subjects. Gene expression levels of UHRF1 and P16INK4A were surveyed using SYBR Green Quantitative Real-time PCR. Statistical analyses were done using SPSS statistical software 21.0.
Results: P16INK4A gene expression showed reduced levels in 80.64% of patients above 45 years of age, while only 32% of patients below 45 years had reduced expression levels. The Spearman correlation test also demonstrated a significant negative correlation between UHRF1 and p16INK4A gene expression levels in AML patients, which was not observed in the control group (r=0.343 and P= 0.015).
Conclusion: Regarding the age-related patterns of UHRF1 and p16INK4A gene expression, and also the presence of negative correlation between them, we conclude that UHRF1 may potentially be involved in p16INK4A down-regulation in elderly AML patients, which may subsequently facilitate the progression of AML in older ages.
Introduction
Acute myelogenous leukemia (AML) is a clonal malignant disease of hematopoietic system, which accumulates in bone marrow and disrupts normal hematopoiesis 1. Despite increased understanding of AML pathophysiology, the mortality rate of AML patients is relatively higher than other hematologic malignancies 2. Besides the disadvantages of current treatment protocols, one reason for poor outcome in AML patients is the absence of an entirely accurate prognostic marker in patients with normal karyotype (which includes nearly 50% of AML patients) 3. Therefore, new studies are necessary for the determination of appropriate prognostic markers and effective treatment protocols. In this regard, recent studies have implicated that epigenetic modifications play a causal role in the initiation and progression of cancer at least as frequently as mutation 45.
Some of the evidence have suggested the prominent pathogenic role of epigenetics in the aggressive behavior of AML versus other genetic lesions 6. For instance, when compared with other human cancers, AML has a paucity of genetic lesions per case 7. Also, recent studies have shown that many AML patients do not carry any recognized AML-associated driver gene mutations 3. Finally, in contrast to solid tumors, sequencing results have shown an infrequent rate of mutations in TSGs in AML patients (except for mutations in WT1 and p53 genes which occur in 10% and 7% of AML patients, respectively) 8.
In comparison with solid tumors, leukemic myeloblasts mainly undergo genome-wide hyper-methylation 9, which represses mainly gene expression of TSGs 10. On the other hand, epigenetic modifications in AML patients have a tendency to induce new mutations 10. The expression of some TSGs is an age-dependent process and increases with aging, probably to prevent clonal expansion of cells that have sufficient number of tumorigenic mutations 1112. In this regard, we studied the expression of p16INK4A, a tumor preventive gene, as it has this pattern of gene expression 13. However, in malignant conditions such as AML, p16INK4A gene expression is reduced with aging, which is mainly due to hyper-methylation of the CDKN2A promoter 14.
Sub-clinical studies have shown that some demethylase agents, such as cladribine and clofarabine, enhance the cytotoxic effect of routine AML therapies by epigenetic modulation of TSGs, such as CDKN2A 15. We also evaluated the gene expression of UHRF1, an oncogene which suppresses the expression of TSGs (such as CDKN2A) in solid tumors 16. UHRF1 is responsible for histone modification (by interacting with G9a, HDAC1 and Suv39H1 proteins) and DNA methylation (by interacting with DNA methyltransferases (DNMTs) to condense the genome and reduce its accessibility to transcription factors 16. On the other hand, UHRF1 marks DNMT1a/3 for proteosomal degradation and, thereby, causes genome-wide hypo-methylation 17.
Regarding genome-wide hyper-methylation 9 and subsequent CDKN2A promoter hyper-methylation 18 in leukemic blasts of AML, to our knowledge, this is the first study to evaluate UHRF1 gene expression level as an important epigenetic regulator and to investigate its correlation with p16INK4B gene expression. These investigations may help us to better understand the processes involved in TSG deregulation in elder AML patients.
Methods
Patients
Peripheral blood (PB) and bone marrow (BM) samples were obtained at the time of diagnosis (prior to cytotoxic chemotherapy) during routine clinical assessment of 50 de novo AML patients. Specimens were collected from all patients with informed consent in agreement with the Declaration of Helsinki and the Ethical Committee of Shahid Beheshti University of Medical Sciences (IR.SBMU.RETECH.REC.1396.800). Diagnosis was made based on PB or BM film examination, immuno-phenotyping, and molecular studies. Immuno-phenotyping analysis was based on the World Health Organization (WHO) classification. Detailed demographic and sub-clinical characteristics of the patient samples are summarized in Table 1. We also collected 18 control samples from PB and BM of healthy persons.
| Study population (N=50) | |
| Age median, y (Range, y)AML M4, M5 | 35 (3-89)6 (12) |
RNA extraction and cDNA synthesis
Mononuclear cells were isolated from PB or BM samples using Ficoll-Hypaque (INTRON, South Korea) density gradient centrifugation. These specimens were immediately cryopreserved or prepared for RNA extraction. Total RNA was extracted from each specimen using 1ml of Trizol. The quantity and quality of total RNA and its contamination with genomic DNA were examined by Nanodrop (Thermo Scientific, USA) and agarose gel electrophoresis. RNA to cDNA conversion was performed according to the cDNA kit (from Thermo Scientific).
Analysis of gene expression by Real-time quantitative PCR (RT-qPCR)
A SYBR Green I Real-time PCR assay was performed in 25 µl final reaction volume using 5 µl cDNA (100 ng RNA equivalent), 0.75 µl primers (300 nM), 12.5 Universal Master Mix (Ampliqon, Denmark), 2.5 µl PCR buffer 10X, and sterile dH2O to reach total volume. Thermal cycling was carried out on Rotor-Gene 6000 (Qiagen, USA) using the following cycling conditions: 10 min at 95oC, followed by 40 cycles at 95 oC for 15 s, and 60 oC for 30 s. Efficiency of all primers was evaluated by triplicate testing of five serial dilutions of cDNA. The fold changes of each gene were calculated by ∆∆CT formula 19.
Statistical analyses
Statistical data were expressed as mean ± SD. All tests were performed in triplicates and standard deviation (SD) less than 0.167 was admissible as a good inter-run reproducibility. Depending on the Shapiro-Wilk test results, we used One-Way ANOVA or Kruskal-Wallis for multi-state variables, and t-test or Mann-Whitney U for two-state variables. For analysis of correlation, Spearman test was used. Two tailed P-value less than 0.05 was considered as significant (Table 2).
| Genes | Primers | Sequences(5'-3') | TM( o C) | Amplicon size (bp) |
| CDKN2A | CDKN2A.F | GCACCAGAGGCAGTAACCA | 59.63 | 101 |
| CDKN2A.R | AGTTTCCCGAGGTTTCTCAGAG | 59.70 | ||
| UHRF1 | UHRF1.F | GCGGGGCTTCTGGTACGAC | 63 | 123 |
| UHRF1.R | TCCACGAAGATGATCCGACAGTC | 62.04 | ||
| ABL | ABLF | TGGAGATAACACTCTAAGCATAACTAAAG | 59.1 | 124 |
| ABLR | GATGTAGTTGCTTGGGACCCA | 60.0 |
Results
Overall, 50 AML patients and 18 healthy volunteers were evaluated in this study. The age of the patients in the study was between 2 -89 years (median 47 years). Patients were divided in three distinct groups, including AML M0-M2 (n=26, 52%), AML M3 (2=18, 36%), and AML M4-M5 (n=6, 12%). Based on immuno-phenotyping parameters (e.g. gate of CD45 dim, the samples ranged from 20-96% (median 80%) blast. The control group included 5 males and 13 females, aged 13-87 years (median 35 years) (Table 1).
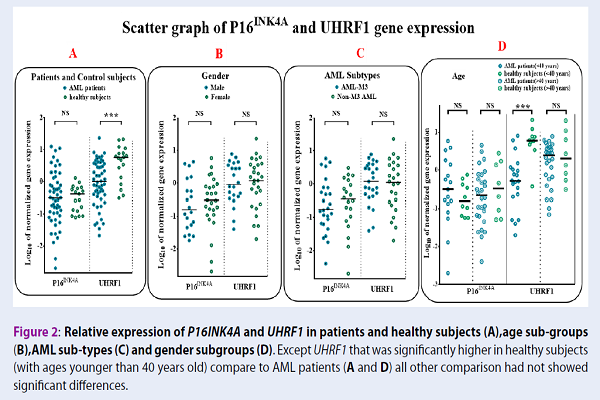
As depicted in Figure 1A, p16INK4A showed increased expression in 13 (26%) of the patients, intermediate expression in 14 (28%) of patients, and a reduced expression in 23 (46%) of patients. In search for criteria that could impact p16INK4A gene expression, we compared gene expression in different sub-groups based on age, gender, blast percent, CD34, HLA-DR positive or negative, and FAB sub-types. We also evaluated the correlation of p16INK4A with continuous variables, including age and blast percent. When comparing any group, no statistically significant difference was observed, except for a negative significant correlation with the age of patients (Figure 2A, r=-0.034, and P=0.016). Based on the regression lines in Figure 2A, 80.64% of patients with age above 45 years showed a decreased expression of p16INK4A, lower than the age-matched control group. However, this situation was observed only in 32% of patients below 45 years. In contrast to AML patients, p16INK4A expression had a positive significant correlation with age in the control group (Figure 2A, control group r = 0.50).
We also evaluated UHRF1 gene expression between different subgroups involved in this study. UHRF1 gene expression was down-regulated in 39 (78%) AML patients, compared with the control group (Figure 1A, Mann-Whitney U test P=0.001). The expression of UHRF1 gene was significantly correlated to age of both normal subjects and AML patients, but in a reciprocal manner (Figure 2B, control group r=-0.45, P=0.05; AML patients r=0.34, P=0.52). Finally, we found that the gene expression of UHRF1 and p16INK4A showed a negative correlation in AML patients, but not in the control group (Figure 2 C, r=-0.343, p=0.015).

Discussion
DNA methylation pattern is commonly an age-dependent process, which is characterized by a drift in CpG island epigenetic marks during aging. This drift gradually causes a distinctive pattern of epigenetic marks in old and young AML patients 1182021. A study showed that the promoter of CDKN2B gene is de-methylated in healthy elder, which causes a higher expression of this gene in older individuals compared with younger ones 20. This pattern of over-expression can induce apoptosis or cellular senescence in cells with genomic damage and is regarded as a protective mechanism against cancer formation 14. However, cancer cells reduce the expression and also function of p16INK4A to overcome this barrier 7. For example, several types of solid tumors increase the expression of UHRF1, an oncogene that recruits methylase enzymes to the CDKN2B promoter for repressing its expression 16.

In contrast with solid tumors, we demonstrated that UHRF1 expression was significantly decreased in 78% of AML patients (39/50 patients). Routinely, UHRF1 over-expression in solid tumors enhances tumor growth and prevents cellular differentiation and senescence 2. UHRF1 overexpression in patients with breast cancer downregulates BRCA1 tumor suppressor and is associated with lower survival rate 21. Various studies have also shown that UHRF1 is over-expressed in other solid tumors such as lung cancer 22, liver cancer 2324, gastric cancer 25, colorectal cancer 2627, and prostate cancer 28. It impacts clinical stage, metastasis, progression, relapse of disease, and overall survival of patients. Compelling evidence have revealed that although UHRF1 over-expression induces global DNA hypo-methylation through DNMT degradation 17, it simultaneously recruits methylase to the genomic regions of TSGs, such as CDH1, P16, P53, P21, KISS1 and PML, and suppresses their expression in malignant conditions 16.
Based on these observations, if over-expression of UHRF1 is useful for malignant cells, it is thereby unclear as to why AML leukemic blasts downregulate UHRF-1 expression. A previous report by Mizuno et al. showed that DNMTs increased in AML patients in comparison with the bone marrow normal cells 19. Further studies revealed that, DNMT enzymes are marked by UHRF1 for future degradation by proteasome system 17. Therefore, regarding our data, UHRF1 down-regulation can be a possible mechanism, underlying DNMT overexpression in AML patients, which is consistent with genomic hypermethylation that occurs in many TSG regions of AML blasts 9. Additionally, other studies on human hepatocellular carcinoma cell lines have demonstrated that UHRF1 deficiency led to an expansion of cancer cells by CXCR4/AKT-JNK/IL-6 signaling pathway activation 29.
Furthermore, we also detected a significant positive correlation between the age of AML patients and UHRF1 gene expression levels (r=0.397 and p=0.005). In our study, patients above 50 years old had a tendency to express UHRF1 in a similar method with healthy subjects, while younger patients had lower levels of UHRF1 in comparison with healthy counterparts. Thus far, no study before ours has investigated UHRF1 gene expression and its correlation with aging.
Consistent with the results obtained from solid tumors 16, in the present study, UHRF1 gene expression had a significant negative correlation with p16INK4A gene expression. Evidence from the evaluation of various types of human cancers, including gastric cancer, colorectal cancer, lung cancer, cervical cancer and pancreatic carcinoma, showed that UHRF1 caused suppression of p16INK4A gene expression through hyper-methylation of its genomic locus 16.
We found that more than 80% of patients who were older than 45 years expressed p16INK4A at a lower level than the normal control group. However, this situation was seen in only 32% of patients below 45 years (on the other hand, 68% of patients below 45 years had p16INK4A over-expression). p16INK4A over-expression in younger patients can be a consequence of their positive regulator over-expression 30 or a physiological response to keep cells from leukemogenesis lesions, as well as preventing occurrence during aging in normal people 14. However, this physiological barrier has some essential defects in elderly AML patients. Consistent with our observations, de Jonge et al. reported that p16INK4A gene expression is reduced by aging in AML patients and impacts overall survival of old patients 931 looking for a reason. We found that old patients who had down-regulation of p16INK4A, express UHRF1 similar to their healthy counterpart. We suggest that a regulated pattern of UHRF1 gene expression is needed to reduce p16INK4A gene expression in AML patients. This regulated pattern not only prevents over-degradation of DNMT enzymes that generally occur in solid tumors (probably due to UHRF1 over-expression) but it can also properly recruit DNMTs to the promoter of CDKN2A gene to suppress its expression.
Conclusions
In this study, for the first time, UHRF1 has been shown to act as a repressor of p16INK4A in elderly AML patients. Down-regulation of p16INK4A may suppress cell physiological defenses against leukemogenesis and dangerous lesions, and may facilitate the development of AML in elderly people.
Competing Interests
The authors declare no conflict of interest.
Authors' Contributions
All authors contributed to the design of the research. VA, MM, MKHF and AGH collected the data. VA, MAF, AH and MM conducted analysis and interpretation of data. All authors drafted the first version. VA, MAF, MM, MKHF, AGH, HA and ZKH edited the first draft. All authors reviewed, commented and approved the final draft.
Acknowledgments
Financial support: Shahid Beheshti University of Medical Sciences, Shahid Beheshti, Iran.
Abbreviations
AML: Acute Myelogenous Leukemia
PCR: Polymerase Chain Reaction
References
-
Khwaja
A.,
Bjorkholm
M.,
Gale
R. E.,
Levine
R. L.,
Jordan
C. T.,
Ehninger
G..
Acute myeloid leukaemia. Nature Reviews. Disease Primers.
2016;
2
:
16010
.
-
Saultz
J. N.,
Garzon
R..
Acute Myeloid Leukemia: A Concise Review. Journal of Clinical Medicine.
2016;
5
:
E33
.
-
Network
Cancer Genome Atlas Research.
Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. The New England Journal of Medicine.
2013;
:
2059-74
.
-
Tsai
C. T.,
So
C. W..
Epigenetic therapies by targeting aberrant histone methylome in AML: molecular mechanisms, current preclinical and clinical development. Oncogene.
2017;
36
:
1753-9
.
-
Baylin
S. B..
DNA methylation and gene silencing in cancer. Nature Clinical Practice. Oncology.
2005;
2
:
S4-11
.
-
Li
S.,
Garrett-Bakelman
F. E.,
Chung
S. S.,
Sanders
M. A.,
Hricik
T.,
Rapaport
F..
Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nature Medicine.
2016;
22
:
792-9
.
-
Alexandrov
L. B.,
Nik-Zainal
S.,
Wedge
D. C.,
Aparicio
S. A.,
Behjati
S.,
Biankin
A. V.,
Australian Pancreatic Cancer Genome
Initiative,
Signatures of mutational processes in human cancer. Nature.
2013;
500
:
415-21
.
-
Patel
J. P.,
Gönen
M.,
Figueroa
M. E.,
Fernandez
H.,
Sun
Z.,
Racevskis
J..
Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England Journal of Medicine.
2012;
366
:
1079-89
.
-
Spencer
David H,
Russler-Germain
David A,
Ketkar
Shamika,
Helton
Nichole M,
Lamprecht
Tamara L,
Fulton
Robert S,
Fronick
Catrina C,
O’Laughlin
Michelle,
Heath
Sharon E,
Shinawi
Marwan.
CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell.
2017;
168
:
801-816. e13
.
-
Hoffbrand
Victor,
Higgs
Douglas R,
Keeling
David M,
Mehta
Atul B.
2016.
Google Scholar -
Wang
Y.,
Zhang
J.,
Xiao
X.,
Liu
H.,
Wang
F.,
Li
S..
The identification of age-associated cancer markers by an integrative analysis of dynamic DNA methylation changes. Scientific Reports.
2016;
6
:
22722
.
-
Juhl-Christensen
C.,
Ommen
H. B.,
Aggerholm
A.,
Lausen
B.,
Kjeldsen
E.,
Hasle
H..
Genetic and epigenetic similarities and differences between childhood and adult AML. Pediatric Blood & Cancer.
2012;
58
:
525-31
.
-
Kim
W. Y.,
Sharpless
N. E..
The regulation of INK4/ARF in cancer and aging. Cell.
2006;
127
:
265-75
.
-
de Jonge
H. J.,
de Bont
E. S.,
Valk
P. J.,
Schuringa
J. J.,
Kies
M.,
Woolthuis
C. M..
AML at older age: age-related gene expression profiles reveal a paradoxical down-regulation of p16INK4A mRNA with prognostic significance. Blood.
2009;
114
:
2869-77
.
-
Valdez
B. C.,
Li
Y.,
Murray
D.,
Ji
J.,
Liu
Y.,
Popat
U..
Comparison of the cytotoxicity of cladribine and clofarabine when combined with fludarabine and busulfan in AML cells: enhancement of cytotoxicity with epigenetic modulators. Experimental Hematology.
2015;
43
:
448-61.e2
.
-
Ashraf
W.,
Ibrahim
A.,
Alhosin
M.,
Zaayter
L.,
Ouararhni
K.,
Papin
C..
The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget.
2017;
8
:
51946-62
.
-
Jia
Y.,
Li
P.,
Fang
L.,
Zhu
H.,
Xu
L.,
Cheng
H..
Negative regulation of DNMT3A de novo DNA methylation by frequently overexpressed UHRF family proteins as a mechanism for widespread DNA hypomethylation in cancer. Cell Discovery.
2016;
2
:
16007
.
-
Clark
S. J.,
Melki
J..
DNA methylation and gene silencing in cancer: which is the guilty party?. Oncogene.
2002;
21
:
5380-7
.
-
Mizuno
S.,
Chijiwa
T.,
Okamura
T.,
Akashi
K.,
Fukumaki
Y.,
Niho
Y..
Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood.
2001;
97
:
1172-9
.
-
Krishnamurthy
J.,
Torrice
C.,
Ramsey
M. R.,
Kovalev
G. I.,
Al-Regaiey
K.,
Su
L..
Ink4a/Arf expression is a biomarker of aging. The Journal of Clinical Investigation.
2004;
114
:
1299-307
.
-
Jin
W.,
Chen
L.,
Chen
Y.,
Xu
S. G.,
Di
G. H.,
Yin
W. J..
UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Research and Treatment.
2010;
123
:
359-73
.
-
Daskalos
A.,
Oleksiewicz
U.,
Filia
A.,
Nikolaidis
G.,
Xinarianos
G.,
Gosney
J. R..
UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer.
2011;
117
:
1027-37
.
-
Wu
S. M.,
Cheng
W. L.,
Liao
C. J.,
Chi
H. C.,
Lin
Y. H.,
Tseng
Y. H..
Negative modulation of the epigenetic regulator, UHRF1, by thyroid hormone receptors suppresses liver cancer cell growth. International Journal of Cancer.
2015;
137
:
37-49
.
-
Mudbhary
R.,
Hoshida
Y.,
Chernyavskaya
Y.,
Jacob
V.,
Villanueva
A.,
Fiel
M. I..
UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell.
2014;
25
:
196-209
.
-
Zhou
L.,
Zhao
X.,
Han
Y.,
Lu
Y.,
Shang
Y.,
Liu
C..
Regulation of UHRF1 by miR-146a/b modulates gastric cancer invasion and metastasis. The FASEB Journal.
2013;
27
:
4929-39
.
-
Wang
F.,
Yang
Y. Z.,
Shi
C. Z.,
Zhang
P.,
Moyer
M. P.,
Zhang
H. Z..
UHRF1 promotes cell growth and metastasis through repression of p16(ink⁴a) in colorectal cancer. Annals of Surgical Oncology.
2012;
19
:
2753-62
.
-
Sabatino
L.,
Fucci
A.,
Pancione
M.,
Carafa
V.,
Nebbioso
A.,
Pistore
C..
UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene.
2012;
31
:
5061-72
.
-
Babbio
F.,
Pistore
C.,
Curti
L.,
Castiglioni
I.,
Kunderfranco
P.,
Brino
L..
The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene.
2012;
31
:
4878-87
.
-
Kim
J. H.,
Shim
J. W.,
Eum
D. Y.,
Kim
S. D.,
Choi
S. H.,
Yang
K..
Downregulation of UHRF1 increases tumor malignancy by activating the CXCR4/AKT-JNK/IL-6/Snail signaling axis in hepatocellular carcinoma cells. Scientific Reports.
2017;
7
:
2798
.
-
Müller-Tidow
C.,
Metzelder
S. K.,
Buerger
H.,
Packeisen
J.,
Ganser
A.,
Heil
G..
Expression of the p14ARF tumor suppressor predicts survival in acute myeloid leukemia. Leukemia.
2004;
18
:
720-6
.
-
Zeighami
Shahryar,
Azizzadeh
Entegham,
Tabatabaee
Hamid Reza,
Adib
Ali,
Babaei
Amir Hossein,
Ariafar
Ali.
Opium and grade of urothelial bladder cancer. Journal of Nephropathology.
2018;
7
(2)
:
69-73
.
Comments
Downloads
Article Details
Volume & Issue : Vol 5 No 9 (2018)
Page No.: 2658-2663
Published on: 2018-09-27
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 4450 times
- Download PDF downloaded - 1275 times
- View Article downloaded - 0 times
